
Overview
The microbiota is the total microbial complex containing a wide range of bacterial species, ranging ubiquitously from humans (e.g., the microbiota inhabiting the animal gut) to the natural environment. In recent years, research on the human gut flora has been conducted worldwide, and the human gut flora has been closely associated with the development of various diseases. Therefore, characterizing the diversity and composition of microbial communities in specimens is one of the main goals of current microbiological research. Effective analysis of microbial communities has historically faced many challenges due to the inability to identify those microorganisms that cannot be cultured and microbial identification analyses have previously been limited to culture-dependent sequencing methods based on Sanger sequencing technology.
With the rapid development of next-generation sequencing (NGS) technologies, metagenomic sequencing is emerging as a powerful approach to understand the complex microbial communities in the human gut. In metagenomic sequencing, the nine highly variable regions (V1-V9) of the 16S rRNA gene are frequently used to determine bacterial classification. Short-read sequencing platforms for reading portions of the 16S rRNA gene are most commonly used, but are unable to produce read lengths that cover the full length of the 16S rRNA gene to account for sequence similarity, and therefore are subject to errors at the species level. Pacific Biosciences (PacBio) and Oxford Nanopore sequencing platforms overcome these technical limitations and are widely used, particularly those affecting read length. CD Genomics offers specialized full-length 16S/18S/ITS amplicon sequencing services to sequence PCR products from specific regions of 16SrDNA.
Full-length 16S Sequencing Technologies
Short-read Sequencing Platforms
Second-generation sequencing technologies analyze microbial diversity by reading the V1, V2, V3, V4, V5, and V6 regions of the 16S rRNA gene. High-throughput short-read sequencing of 16S rRNA gene amplicons based on the Illumina MiSeq 2 × 300 bp platform specifically targets the highly variable V3-V4 region of the nine variable regions. By reducing the high-cost burden of NGS, it is widely used in various metagenomic studies. However, this short-read amplicon-based platform is not only susceptible to identification bias from potential chimeric sequences generated during PCR amplification for library construction but also limited to genus-level microbial classification based on commonly used 16S rRNA genes based on microbial taxonomic databases. Therefore, the amplicon approach for partially variable regions (V3-V4) is limited to strains with a high degree of similarity at the species level.
Long-read Sequencing Platforms
In microbiome research, this long-read sequencing technology has led to changes in the analysis of complex microbial communities by addressing the problem of identification accuracy that occurs when reading partially highly variable regions of 16S rRNA genes.
The development of nucleic acid sequencing technology has been revolutionized with the introduction of the MinIONTM, a state-of-the-art portable sequencer designed by Oxford Nanopore Technologies. This device, although compact in size, has brought about a profound shift in our approach to sequencing the full-length 16S rRNA gene, which is the cornerstone of bacterial identification and classification studies. The advantages of the MinIONTMare primarily in its comprehensive targeting mechanism, which captures the entire spectrum of the 16S rRNA gene, thus avoiding the pitfalls of fragment sequencing. In this way, it improves the fineness of identification of bacterial species and subspecies, thus enhancing our diagnostic and research capabilities to an unprecedented level. In addition, the specificity of the MinIONTM method ensures a dramatic increase in sensitivity and accuracy, both of which are critical in microbiology research, where even small differences can represent very different bacterial taxa.
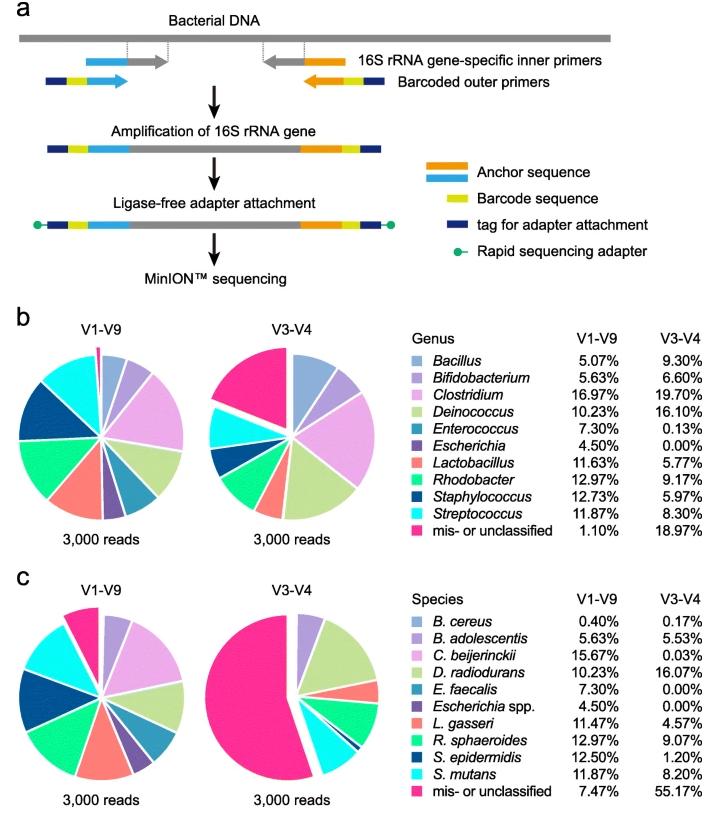
16S rRNA gene sequence analysis using the MinION™ nanopore sequencer. (Matsuo et al., 2021)
Pacific Bioscience SMRT sequencing technology enables accurate sequencing of the entire 16S rRNA gene, especially when delving into the implications of covering all variable regions. Typically, the read length provided by a sequencing platform is known to affect either accuracy or sequence length. However, PacBio introduces an innovative balance between these two parameters. Their high fidelity (HiFi) reads average approximately 1.5 kb in length, ensuring that the majority of genomic information is retained and accurately represented. A key factor driving PacBio's sequencing proficiency is the utilization of the Cyclic Coherent Sequencing (CCS) mode. Through this mechanism, sequencing cycles are repeated multiple times on a single molecule, improving overall sequence accuracy by integrating the data obtained in these cycles. As a result, PacBio HiFi reads offer a dual advantage - superior length and unrivaled accuracy. This combination raises the detection of microbial taxonomic units to previously unattained levels of granularity and precision, with the potential to revolutionize our understanding of microbial ecosystems.
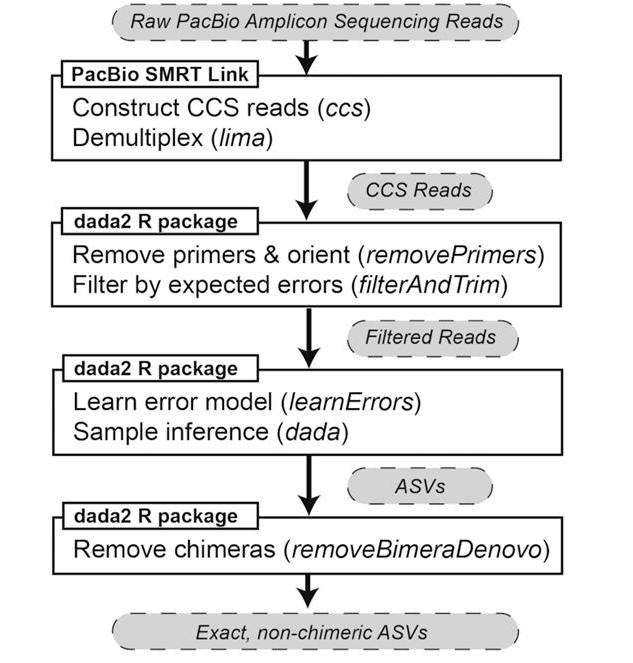
Two software packages, PacBio SMRT Link and the DADA2 R package, were used to process raw PacBio amplicon sequencing data into chimera-free amplicon sequence variants (ASVs). (Callahan et al., 2019)
Synthetic Long Read Length Technology
Long-read sequencing platforms generate reads with lower nucleotide accuracy than Illumina platforms (~15% versus ~0.1% for Illumina) due to random base detection errors that occur during multiple sequencing of the same region. Loop Genomics has introduced a new 16S full-length-based synthetic long-read (sFL16S) technology. The method represents a hybrid approach that combines the reliability of short-read long sequencing with the depth and detail of long-read sequencing. The technology reads the entire variable region of the 16S rRNA gene (V1-V9) to elucidate microbial communities in metagenomic studies. With unique molecular barcoding technology, fragmented short read lengths are assembled into individual full-length 16S rRNA genes, resulting in significantly fewer false positives and higher classification accuracy.
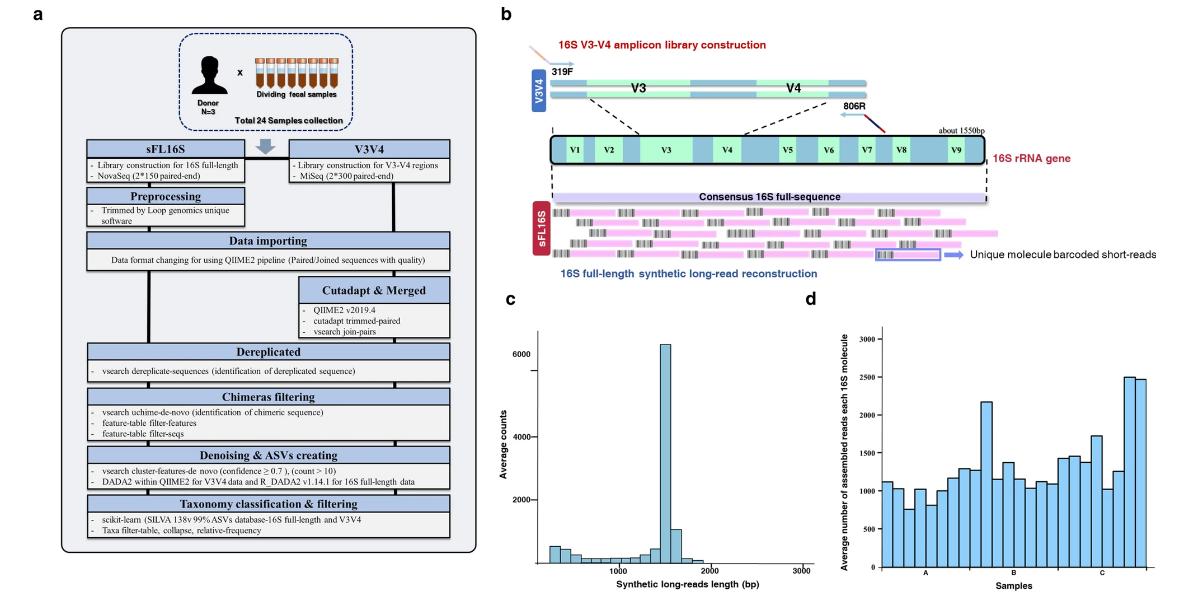
Technical introduction and analysis workflow for a new 16S full-length-based synthetic long-read (sFL16S) technology. (Jeong et al., 2021)
Applications of Full-length 16S rRNA Sequencing
Characterization of microbial communities
The ability to sequence full-length 16S rRNA genes provides a panoramic view of microbial taxa in a sample, paving the way for detailed community characterization.
Microbial biomarker detection
With the increased resolution provided by full-length sequencing, specific microbial signatures or biomarkers indicative of particular conditions or environmental factors can be more accurately identified.
Disease surveillance
Changes in microbial communities that can be discerned through full-length sequencing can serve as indicators of the onset, progression, or resolution of certain diseases, particularly human intestinal diseases.
Drug development
Analyzing microbial communities at high resolution can help develop therapies that target specific microbial pathogens or modulate microbial communities for beneficial effects.
References
- Matsuo, Yoshiyuki, et al. "Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION™ nanopore sequencing confers species-level resolution." BMC microbiology 21 (2021): 1-13.
- Callahan, Benjamin J., et al. "High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution." Nucleic acids research 47.18 (2019): e103-e103.
- Jeong, Jinuk, et al. "The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology." Scientific reports 11.1 (2021): 1727.













