
Overview of DAP-seq
The DNA Affinity Purification Sequencing (DAP-seq) technique is an innovative method aimed at exploring the interactions between transcription factors (TFs) and genomic DNA. By simulating the interaction between TFs and their DNA binding sites in vitro, this technology reveals key DNA sequences in gene regulatory networks. In contrast to traditional Chromatin Immunoprecipitation Sequencing (ChIP-seq), DAP-seq is not limited by antibody availability or species constraints, providing researchers with a more flexible research approach.
DAP-seq cleverly combines in vitro protein expression with high-throughput sequencing technology, eliminating the need for specific antibodies for each target TF. This approach allows for the comprehensive identification and analysis of transcription factor binding patterns throughout the entire genome in an environment unaffected by cellular chromatin structure. This is crucial for gaining deeper insights into complex transcriptional regulatory networks and their impact on organismal growth, development, and environmental adaptability.
Through DAP-seq, researchers can more precisely elucidate the molecular mechanisms of gene expression regulation and how environmental factors, such as DNA methylation, regulate these processes. The application of this technology not only advances our understanding of gene regulation mechanisms but also provides new perspectives and tools for future biomedical research and potential therapeutic strategies.
Principles of DAP-seq
The cornerstone of DAP-seq is the replication of TFs naturally interacting with DNA within the cellular environment, albeit performed ex vivo. This groundbreaking methodology empowers researchers to directly observe and decipher TFs' genomic binding sites, unaffected by the complexities of the intracellular milieu. Central to DAP-seq's essence is the employment of an ex vivo expression system to engineer TFs fused with affinity tags. These tagged TFs are subsequently utilized to precisely isolate specific genomic binding sites through affinity purification techniques. Through meticulous examination of sequencing data, researchers can accurately pinpoint the genomic regions where TFs bind, thereby facilitating a comprehensive exploration of the intricate landscape of gene regulatory networks.
Workflow of DAP-seq
The DAP-seq methodology stands as a robust technique for discerning the binding sites of TFs throughout the entirety of the genome. This approach seamlessly integrates principles from molecular biology, biochemistry, and high-throughput sequencing technologies. Below is an elucidation of the meticulous steps constituting the DAP-seq workflow:
Genomic DNA Library Construction
- Sample Preparation: Initially, genomic DNA of high quality is extracted from the target organism, ensuring pristine purity devoid of protein or RNA contamination.
- DNA Fragmentation: The extracted genomic DNA undergoes physical fragmentation, typically yielding fragments ranging from 200 to 500 base pairs, facilitated by methods such as sonication or enzymatic digestion.
- End Repair and A-Tailing: Fragmented DNA is subject to end repair to generate blunt ends compatible with adapters. Subsequently, an enzymatic reaction appends an adenine nucleotide (A-tail) to the 3' end of each DNA fragment.
- Adapter Ligation: Double-stranded DNA adapters harboring essential sequences for PCR amplification and sequencing are ligated to both ends of the DNA fragments, facilitating the construction of the genomic DNA library.
In Vitro Protein Expression
- Expression of TFs: The coding sequences (CDS) encoding target TFs are cloned into expression vectors bearing affinity tags like HaloTag. In vitro expression systems, such as wheat germ extracts or rabbit reticulocyte lysates, are employed for the expression of tagged TFs.
Affinity Purification and Sequencing
- Purification of TFs: Affinity chromatography utilizing magnetic beads is employed to purify tagged TFs from the expression system.
- TF-DNA Binding: Purified TFs are incubated with the adapter-ligated genomic DNA library, allowing them to bind to DNA fragments at their respective binding sites.
- Non-specific DNA Washing: Multiple washing steps are performed to eliminate non-specifically bound DNA fragments and proteins, thereby enhancing result specificity.
- DNA Elution and Recovery: DNA fragments bound to TFs are eluted from the magnetic beads, typically achieved by denaturing proteins through heating, thereby releasing bound DNA.
- PCR Amplification: The eluted DNA undergoes PCR amplification to augment the requisite DNA quantity for sequencing. Index sequences are introduced during this step for sample multiplexing.
- Validation and Quantification of Library: Library concentration is determined via fluorescence quantification methods like Qubit. PCR is employed to validate the correctness of adapter ligation.
- Sequencing: Prepared libraries are subjected to sequencing on high-throughput platforms, predominantly utilizing Illumina platforms for single-end or paired-end sequencing.
Data Analysis
- Data Analysis: Raw sequencing data undergoes stringent quality control, filtering, alignment to the reference genome, peak calling, and motif identification to delineate TF binding sites and potential DNA binding motifs.
- Result Interpretation: The binding characteristics and functions of TFs are interpreted based on peak distribution, motif analysis, and existing biological knowledge. Comparative analysis with extant ChIP-seq data or other epigenetic modification data aids in elucidating the role of TFs in gene regulation.
- The DAP-seq workflow emerges as an indispensable tool for molecular biologists unraveling the intricacies of gene expression regulation. Its provision of a high-resolution view of TF binding events finds multifarious applications in the domains of genomics and systems biology.
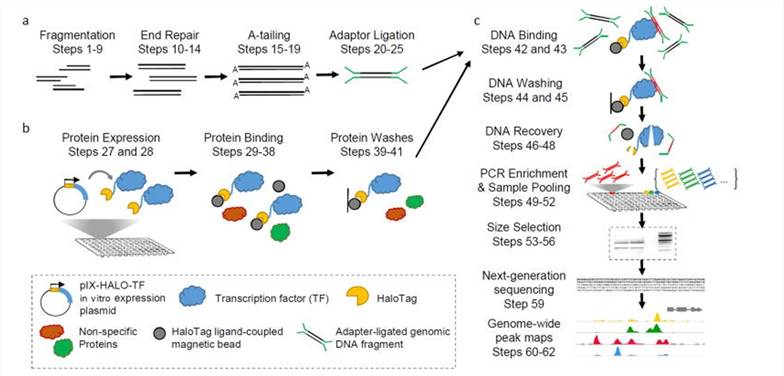 DAP-seq protocol overview(Bartlett et al., 2017)
DAP-seq protocol overview(Bartlett et al., 2017)
Bioinformatics analysis
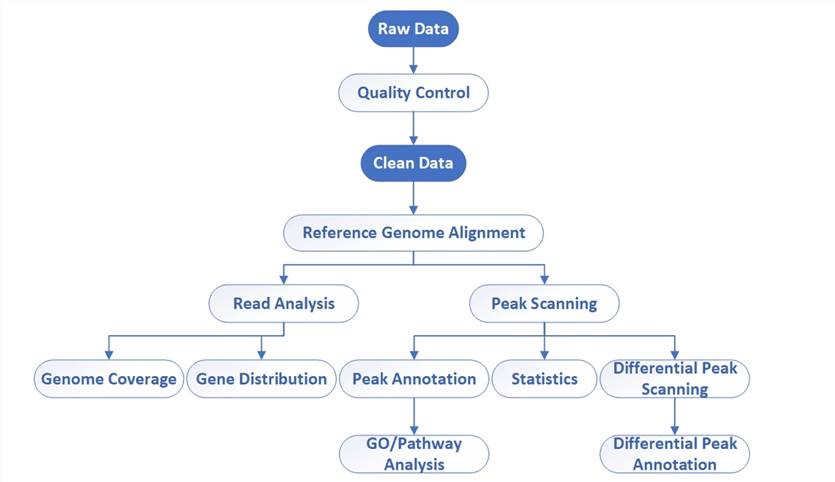
Bioinformatics workflow of DAP-Seq
- Quality Control: Evaluate the quality of raw data using software such as FastQC to ensure accuracy and usability.
- Clean Data: Process the raw data based on the results of quality control, removing adapters and filtering out low-quality reads and possible sequencing errors to improve data quality.
- Reference Genome Alignment: Align cleaned FASTQ files to a reference genome using Bowtie2, BWA, or other alignment tools to determine the location of reads in the genome.
- Peak Calling: Identify peak regions of transcription factor binding on the genome, typically manifested as areas of enriched reads.
- Differential Peak Analysis: Identify peak regions significantly different between samples under different conditions, if comparing different conditions.
- Genome Coverage: Evaluate the coverage range of TFs in the genome and the distribution of peak regions across the entire genome.
- Gene Distribution: Analyze the relationship between peak regions and genes to determine genes that TFs may regulate.
- Peak Annotation: Map peak regions to functional regions of the genome, such as promoters, enhancers, etc., and conduct sequence motif analysis.
- Statistical Analysis: Analyze statistical characteristics of peak regions, such as size, intensity, and significance.
- GO/Pathway Analysis: Associate potential target genes with gene ontology and biological pathway databases to infer the biological function of TFs.
- Differential Peak Annotation: Provide detailed functional annotations for differential peaks to reveal the regulatory mechanisms of TFs under different conditions.
CD Genomics offers comprehensive and accurate DAP-seq technology services, with experienced expert teams performing quality control at each step of the process to ensure result accuracy.
Service you may intersted in
Advantages and limitations
DAP-seq, an innovative method for probing the intricate dance between proteins and DNA, emerges as a formidable asset in the quest to map TF binding sites across the vast expanse of the genome. Its prowess lies in its precision, capable of pinpointing TF binding sites with remarkable accuracy across the entire genomic landscape, thereby offering a panoramic view of TF DNA binding specificity. For instance, researchers have harnessed the power of DAP-seq to chart a comprehensive map of TF binding sites throughout the Arabidopsis genome, shedding light on how DNA methylation influences TF binding dynamics. In comparison to traditional ChIP-seq approaches, DAP-seq dazzles with its heightened resolution, facilitating the precise delineation of TF binding regions sans the need for specific antibodies, thus alleviating concerns regarding antibody quality and nonspecific binding. Moreover, DAP-seq transcends the realm of TFs, delving into the binding characteristics of a myriad of DNA-binding proteins, thereby expanding its utility in unraveling molecular mysteries.
However, notwithstanding its advantages in identifying TF binding sites, DAP-seq grapples with certain limitations. The preparation of top-tier protein and DNA samples presents challenges, particularly when contending with proteins resistant to purification. Furthermore, despite its exceptional resolution, the specter of false positives and negatives looms, especially in the detection of low-abundance binding sites, resulting in variable success rates in identifying binding sites for different TF families. Additionally, the copious data output from DAP-seq necessitates sophisticated bioinformatics tools and expertise for robust data analysis and interpretation. Moreover, fine-tuning of DAP-seq protocols may be imperative for different proteins and biological samples to achieve optimal detection of binding sites.
In summation, while DAP-seq stands as a potent instrument for unraveling the regulatory ballet orchestrated by TFs in governing gene expression, its effective deployment mandates meticulous sample preparation, rigorous data analysis, and potential refinements in technique.
References
- O'Malley RC, Huang SC, Song L, et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape [published correction appears in Cell. 2016 Sep 8;166(6):1598]. Cell. 2016;165(5):1280-1292. doi:10.1016/j.cell.2016.04.038
- Bartlett A, O'Malley RC, Huang SC, et al. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat Protoc. 2017;12(8):1659-1672. doi:10.1038/nprot.2017.055













